Computational Biology Research Center[CBRC]
Advanced Industrial Science and Technology[AIST]| Key Word Search |

|
- Top
- >
- News & Topics
- >
- 2004/12/24
News & Topics
CBRC Researchers Won the Third and the Fourth Prizes at CASP 6
updated on 24th Jan. 2006
Two teams from the Computational Biology Research Center(CBRC) have been awarded the third and the fourth prizes in CASP 6, a protein structure prediction Olympics held in the summer 2004.
About CASP [ Link to CASP 6 website]
Critical Assessment of Techniques for Protein Structure Prediction (CASP) is a bi-annual international contest in which registered research teams respond with their protein structural models to questions provided on the Internet. For the sixth session this year, 87 targets (number of proteins) were given from June 7th to September 2nd. More than 200 teams worldwide competed with their Protein Structure Prediction Techniques.
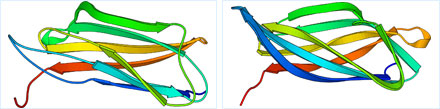
(Target No.212 in CASP6)
Sor45 Protein Predicted Structure (left) and the Native Structure (right)
Our teams were able to succeed in this difficult target by template based methods.
Records of the Two Teams participated from the CBRC
The 3rd Prize in the “Fold Recognition Division” for the team of Kentaro Tomii and his colleagues; and the 4th Prize in “Domain Prediction Division” for the team led by Tamotsu Noguchi were awarded.
In the fold recognition division, participants competed to find similarities of protein structures to those of known protein structures and to construct those models by using sequence homology analysis and structural recognition techniques while raising the analysis sensitivity. The team of Tomii and others employed “FORTE-SUITE” - a system developed based on unique structural recognition technology “FORTE” - and won the third place. The team was invited to Italy to give a presentation of their achievements from December 4th to December 8th. The prized visiting public lecture was the first time in six years since when the team of National Institute of Genetics precented in CASP 3.In addition, it is not an exaggeration to say that our technology is the best in the world as a single technique because the top two teams used Meta-Server, with which the teams used secondary information provided from other teams.
The FORTE-SUITE system used in this contest is a technique to predict protein structures. It does so by repeating trial-and-error based side-chain modeling based on multiple candidate structures that are selected with accurate alignment method (FORTE Method; Patent Pending) which utilizes the correlated coefficient as a similarity measurement between two profile columns to be compared.
In the near future, these techniques developed by our teams are expected to be applied to predict protein structures and functions for genes whose information will become more available through the Genome Analysis Project.